Determination of Sodium Propionate in Whole Tobacco
Health Canada
T-312 December 31, 1999
Table of Contents
- Scope of Applications
- Normative References
- Definitions
- Method Summary
- Apparatus and Equipment
- Reagents and Supplies
- Preparation of Glassware
- Preparation of Solutions
- Preparation of Standards
- Sampling
- Sample Analysis
- Quality Control
- References
- Appendix
1 Scope of Applications
- This method describes the extraction of sodium propionate in whole tobacco and its quantification by GC with capillary column technology and flame ionization detection.
- This method is to be used to determine the amount of Sodium Propionate added to tobacco as a mold preventative or fungicide. The method is designed to be used as a routine analysis without the need for derivitization.
- This method does not distinguish between the amount of Sodium Propionate added and the amount of naturally occurring propionic acid (if any) found in whole tobacco. This method does not distinguish between Sodium Propionate and propionic acid that may result from some forms of anaerobic fermentation.
- This method is not intended to be used for trace analysis, but is to be used for determining Sodium Propionate that is used in an additive range up to 0.4 %.
2 Normative References
- Health Canada Test Method T-115 - Determination of Tar, Water, Nicotine and Carbon Monoxide in Mainstream Tobacco Smoke, 1999-12-31.
- American Society for Testing and Materials (ASTM) D 1193-77 - Standard Specification for Reagent Water, Version 1977.
- Health Canada Test Method T-402 - Preparation of cigarettes, cigarette tobacco, cigars, kreteks, bidis, packaged leaf tobacco, pipe tobacco, and smokeless tobacco for testing, 1999-12-31.
3 Definitions
- Refer to T-115 for definitions of terms used in this document.
4 Method Summary
- Whole tobacco is extracted in a 20 % aqueous solution of 0.01N H2SO4 in methanol under extremely gentle conditions.
- Calcium chloride (CaCl2) is used to saturate the ionic species of the solution. The propionate ion's equilibrium with the free acid is quantitatively forced toward the free acid as a consequence of its small Ka value (1.34 × 10-5 ).
- Iso-butyric acid (Ka = 1.44 × 10-5) is used as an internal standard. (It also has the effect of minimizing the potential pH differences between samples and standards, which may affect the ion: free acid ratio).
- The free acid is then analyzed by gas chromatography using a splitless injection of the methanolic solution onto a Stabilwax-DA fused silica capillary column (or equivalent) with a flame ionization detector (FID).
- Quantification is achieved by internal standard calibration procedures where the relative response of the samples is compared against a five point calibration of the corresponding acid of the same type of solution.
Note: The testing and evaluation of certain products against this test method may require the use of materials and or equipment that could potentially be hazardous and this document does not purport to address all the safety aspects associated with its use. Anyone using this test method has the responsibility to consult with the appropriate authorities and to establish health and safety practices in conjunction with any existing applicable regulatory requirements prior to its use.
5 Apparatus and Equipment
- 125 mL Erlenmeyer flasks - Polymethylpentene (PMP) with screw Top.
- 2000 mL volumetric flask.
- 100 mL volumetric flask.
- 100 mL graduate cylinder.
- 16 × 25 mm screw-top culture tubes with caps (without liners).
- Finnpipette, 0 - 5 mL or equivalent.
- Micropipettes for the preparation of analytical standards.
- Dispensette, 10 - 50 mL.
- 200 µL micropipette.
- 25 µL micropipette.
- Gas Chromatograph with:
- Splitless, Capillary Injector.
- Autosampler.
- Flame Ionization Detector.
- Data collection system.
- Autosampler vials/caps/Teflon faced septa.
- Stabilwax-DA 30 m × 0.32 mm × 1.0 µm (Crossbond Carbowax - PEG for acidic compounds) or Nukol (30 m X 0.32 mm X 0.25 µm) or equivalent.
- Centrifuge.
- Vortex.
- Lyophilizer.
- Wrist Action Shaker.
6 Reagents and Supplies
Note: All reagents shall be, at the least, recognized as analytical reagent grade in quality.
- Sodium Propionate (NaProp).
- Iso-Butyric Acid.
- Sulfuric acid (H2SO4) 95 - 97 %.
- Type I water - as specified in ASTM D1193.
- Methanol.
- Calcuim Chloride (CaCl2).
- Optional Reagents (for potential quantitation of other organic acids):
- n-Butyric Acid.
- Sodium Acetate.
- Parafilm® or equivalent.
7 Preparation of Glassware
- Glassware should be cleaned and dried in such a manner to ensure that contamination from glassware does not occur.
8 Preparation of Solutions
- Tobacco Extraction Solution (Aqueous Component)
- Add 35 of CaCl2 to a 2000 mL volumetric flask.
- Add approximately 1 L of Type I water and mix until all CaCl2 is dissolved.
- Rinse the neck of the flask to wash down high concentration points of CaCl2. If a white precipitate forms, re-make the solution taking better care of washing down the neck of the flask with type I water.
- Add 0.54 mL of Concentrated H2SO4.
- Make to volume with Type I water.
- 0.01N H2SO4
- Add 0.27 mL of concentrated H2SO4 to approximately 700 mL Type I water in a 1 L volumetric flask.
- Make up to volume with Type I water.
9 Preparation of Standards
- Sodium Propionate Primary Standard (Final concentration approximately 20 mg/mL)
- Accurately weigh 2 g of sodium propionate into a 100 mL volumetric flask.
- Make to volume with 0.01N H2SO4.
- Iso-Butyric Acid Standard- ISTD (Final concentration approximately 100 mg/mL)
- Accurately weigh 2.5 g of iso-butyric acid into a 25 mL volumetric flask.
- Make to volume with aqueous 0.01N H2SO4.
- Working standards
- All standards are made to 25 mL in volumetric flasks with methanol.
- An attempt to maintain a 10 % aqueous fraction for each of the standards is made by adding the appropriate amount of aqueous extraction component (to the nearest 0.05 mL). This is required to maintain a constant aqueous: organic ratio between standards and samples.
This table displays the standard identification for blank 0, and standard 0 to 5. The table includes the volume of primary sodium standard, internal standard added and extraction solution for each standard. Furthermore, it also includes the sodium propionate concentration for each standard.
Standard
IDVolume (µL)
of 1o Na
Prop. StdVolume
(µL) of
Iso-But.
(ISTD)Volume (mL)
Extraction
SolutionNa Prop.
(µg/mL)Blk 0 0 25 2.50 0 Std 1 20 25 2.50 16 Std 2 50 25 2.45 40 Std 3 125 25 2.40 100 Std 4 200 25 2.30 160 Std 5 300 25 2.20 240
Note 1: The concentration of sodium propionate will vary depending on the exact concentration of primary stock prepared.
Note 2: The concentration of ISTD is approximately 100µg/mL (again depending on the exact concentration of the primary stock).
10 Sampling
- The sampling of tobacco products for the purpose of testing shall be as specified in T-115.
11 Sample Analysis
- Extraction of Whole Tobacco
- Accurately weigh 10 g of tobacco ("as is") into a 125 mL PMP Erlenmeyer flask.
- Add 20 mL of the Aqueous Extraction Component to the sample.
- Add 200 µl of iso-butyric acid stock as an internal standard (using micropipette).
- Moisten the tobacco by rotating the flask until all the solution is absorbed by the tobacco or the tobacco appears to be saturated.
- Add 80 mL of methanol to each of the samples.
- Close the flask (seal with Parafilm® if necessary) and shake flask for 30 minutes.
- Let the samples sit for a minimum one hour after shaking.
- Shake each sample for an additional 30 seconds after letting the samples sit.
- Decant the liquid into a 16 X 25 mm screw top culture tube and cap so that none of the solution is allowed to evaporate.
- Sample Clean-up
- Allow the precipitate to settle in the culture tube by letting it sit for two hours. Alternatively: Centrifuge at low speed to settle the precipitate.
- Quantitatively transfer 2 mL of the supernatant to a second 16 X 25 mm screw top culture tube.
- Quantitatively add 2 mL of methanol to each of the aliquots and cap each tube.
- Vortex each of the samples for 10 seconds at high speed.
Note: The remainder of the supernatant should also be vortexed and stored in the refrigerator in the event the sample may require to be repeated. - Centrifuge the diluted sample at medium speed to precipitate any salts that may have formed in the dilution step
- Transfer the supernatant to an autosampler vial to analyze on the GC.
- Instrument Analysis: Gas Chromatograph Configuration
- Injector: Splitless with purge delay of 0.10 minutes.
- Deactivated Splitless Insert.
- Detector: Flame Ionization (FID).
- Carrier: Heat 12.0 psi, flow = 2.3 mL/minute @ 70 °C.
- Split (purge flow) = 20 mL/minute.
Note: Detector gas flows set as per manufacturer's specification
Note: The Stabilwax-DA is specifically deactivated to not react with acidic compounds. For this reason the free acid can be analyzed with minimal tailing effects caused by reactivity with the column.
Note: Methanol causes an enormous amount of tailing. This effect is minimized by having a very short purge delay (relay time) and using a relatively low injector temperature. Ideally, a split injection practically eliminates the tailing, however, this drastically reduces the sensitivity of the method.
- Gas Chromatograph Operating Conditions
- Injector: 225 °C.
- Detector: 230 °C.
- Column Oven Temperature Profile:
Start Temperature: 70 °C Hold for two minutes.
Rate: 7.5 °C/minute to 155 °C Hold for 2.67 minutes.
Rate: 7.5 °C/minute to 205 °C Hold for 4.0 minutes.
Total Run Time: 25.00 minutes.
Temperature programming of the column may require slight changes as the column conditions change after repeated use.
- Autosampler Conditions: Injection volume: 1.0 µL.
- Calculations
- All calculations are performed using an internal standard calibration response factor from the area counts of the standard solutions.
- These calibration response factors are used to calculate the µg/mL concentration of each analyte in the sample, which in turn, with the appropriate multiplier (total volume in mL) and divisor (original sample weight in g) are used to calculate the concentration of Sodium Propionate in the sample.
- To convert concentration to a percentage (%), the [µg/g] result must be divided by 10000.
- All results are expressed on an "as received" basis. These may be expressed on a dry matter basis using the appropriate moisture result.
- Representative Calculations are as follows:
- Analytical Result
NaProp (μg/g) = [Area of Analyte Sample / Area ISTD Sample] X [RF μg/mL] X [Multiplier (mL) / Divisor (g)]
where RF is defined from the calibration curve (or as the concentration per unit area). - Conversion to Percent (as is)
NaProp (%) as is = Result in μg/g /10000
- Conversion to a Dry Matter Basis
NaProp (%) dry matter = NaProp (%) as is / (1- (% Moisture))
where the % Moisture is determined from the same sample as received for sodium propionate analysis.
- Analytical Result
12 Quality Control
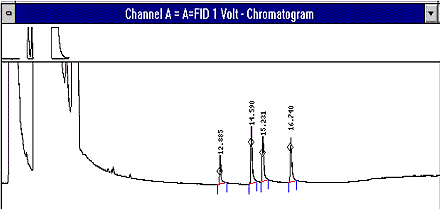
- Typical Chromatogram
- see Figure 1
- Recoveries and Levels of Contamination
- Each analytical run of test cigarettes should also include:
- A Laboratory Reagent Blank (LRB) to evaluate the extent of any interference due to glassware, reagents, and analyzer effects.
- A Laboratory Fortified Blank (LFB) to evaluate the extent of potential analyte loss.
- A Laboratory Fortified Matrix (LFM) to assess matrix interference. This is accomplished by spiking a true sample with a known concentration and determining a per cent recovery.
- Each analytical run of test cigarettes should also include:
- Method Detection Limit (MDL) and Limit of Quantitation (LOQ)
- Method Detection Limit (MDL)
- The method detection limit is determined by analyzing the lowest level standard at least 10 times as an unknown over several days. The MDL is then calculated as three times the standard deviation of these determinations.
- Limit of Quantitation (LOQ)
- The limit of quantification is determined by analyzing the lowest level standard at least 10 times as an unknown over several days. The LOQ is then calculated as 10 times the standard deviation of these determinations.
- Method Detection Limit (MDL)
- Stability of Reagents and Supplies
- All primary stock Sodium propionate standards are prepared fresh weekly.
- All work standards, and extraction solvents are prepared fresh weekly.
- All samples are analyzed as soon as possible after extraction (within 24 hours).
13 References
- AOAC Official Method 971.11, Fatty Acids (Volatile) in Eggs, Gas Chromatographic Method, AOAC Official Methods of Analysis, 1995, Ch. 34, p. 10-12.
- CRC Handbook of Chemistry and Physics, 58th Edition, CRC Press Inc. 1977- 1978, p. D-150, D-151.
- Determination of Sorbic Acid in Tobacco, RJR Macdonald R & D, p. 56-60.
- Organic Chemistry, 2nd Edition, Fessenden and Fessenden, Willard Grant Press, 1979, p. 588, 627, 628.
- The Merck Index, An Encyclopedia of Chemicals and Drugs, 8th Edition, Merck & Co. Inc., 1968.
Appendix
Figure 1: Example Chromatogram of highest organic acid standard. Order of elution is acetic acid, propionic acid, iso-butyric acid, and n-butyric acid.
This figure shows an example chromatogram of the highest organic acid standard. The order for elution is acetic acid, propionic acid, iso-butyric acid, and n-butyric acid.